

Abstract
Background: Chronic lymphocytic leukemia (CLL) development is associated with global immunodeficiency including T-cell exhaustion. We hypothesise that repairing T cell functions would improve outcome and decrease infectious complications which cause significant morbidity in CLL patients. Chronic B-cell receptor (BCR) activation as well as close interactions with the tumour microenvironment promote survival of malignant CLL B-cells, supporting their ability to induce immune suppression. To date, the most clinically successful approach to BCR-signalling inhibition is by the use of BTK inhibitors (BTKi). It has been suggested that the BTKi Ibrutinib has the ability to modulate T-helper cell polarity from Th2 to Th1 and thus would be a step towards repairing CLL associated T-cell defects (1). We were therefore interested to determine whether the second generation BTKi Acalabrutinib which has no reported inhibitory capacity towards ITK would have similar effects than Ibrutinib in modulating T cell responses.
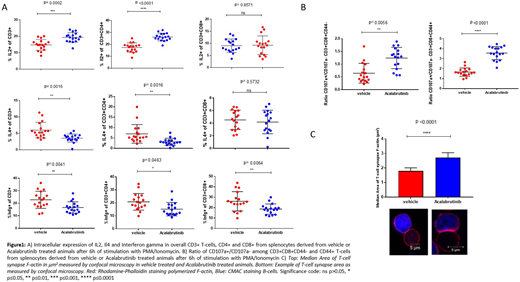
Materials and Methods: To address this question in vivo C57/Bl6 animals 2.5 months of age were injected with 40x10e6 purified CLL B-cells pooled from Eµ-TCL1 mice with CLL. When peripheral blood CLL load reached >10% animals were randomized (mean day 14) to either vehicle treatment (2% HPBD) or Acalabrutinib treatment (0.15 mg/l in 2% HPBC) for 21 days. 17 animals were group were analysed. Splenic cells were isolated, the cellular component characterized by CyTOF and T cell function assessed by multi-parameter flow cytometry and T-cell synapse formation assay.
Results: Treatment with Acalabrutinib resulted in increased expression of IL2 (p<0.0001) in CD4+ T cells and decreased expression of IL4 among both CD4+ T cells (p=0.0016) but not CD8+ T-cells. There was a reduction in Interferon gamma production in both CD4 T-cells (p=0.0463) and CD8+ T-cells (p=0.0064) with Acalabrutinib treatment. In addition, treatment resulted in an increase in CD107a+/CD107a- ratio among both CD44+ and CD44- CD8+ cytotoxic T-cells. This effect was pronounced in the antigen experienced CD44+ cytotoxic T-cells (p<0.0001) but only moderate (p=0.0056) in the CD44- cytotoxic T-cells. Lastly, we find a statistically significant increase in T-cell synapse area (p<0.0001) with Acalabrutinib treatment (Figure 1).
Conclusion: We find that treatment with both Ibrutinib and Acalabrutinib result in a similar shift of T cell function with cytokine secretion with increased IL2 and decreased IL4. T-cells in CLL have increased Interferon gamma production (2) and the observed decrease seen with Acalabrutinib is in keeping with a normalization of T cell function. Moreover, overall CD8+ T-cell function is increased with Acalabrutinib treatment as evidenced by an increase in cytotoxic T-cell function and immune synapse formation. We speculate that inhibition of ITK is not the leading cause for this phenomenon as Acalabrutinib does not have inhibitory capacity toward this kinase. These changes suggest that BTKi modulate T cell mediated immune responses indirectly via either their effects in the CLL B-cell or myeloid cells in the tumour microenvironment.
References
Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539-49.
Riches JC, Davies JK, McClanahan F, Fatah R, Iqbal S, Agrawal S, et al. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood. 2013;121(9):1612-21.
Frigault:Acerta Pharma: Employment. Gribben:Abbvie: Honoraria; Acerta Pharma: Honoraria, Research Funding; Janssen: Honoraria, Research Funding; Wellcome Trust: Research Funding; Celgene: Consultancy, Honoraria, Research Funding; Unum: Equity Ownership; Roche: Honoraria; TG Therapeutics: Honoraria; NIH: Research Funding; Medical Research Council: Research Funding; Cancer Research UK: Research Funding; Kite: Honoraria; Pharmacyclics: Honoraria; Novartis: Honoraria.

